隨著人類基因組計劃的完成和測序技術的不斷發展,單細胞測序技術已應用于多個生物醫學領域,且在分析腫瘤細胞亞群、揭示細胞異質性和研究腫瘤的發生、演變及耐藥性等方面具有獨特的優勢。前不久給大家介紹過單細胞數據分析中的細胞通訊分析,今天要為大家介紹惡性腫瘤分析。

inferCNV 是 broad 研究所開發的用來探索腫瘤單細胞 RNA-seq 數據的軟件,分析其中的體細胞大規模染色體拷貝數變異(copy number alterations, CNA), 比如整條染色體或大片段染色體的增加或缺失(gain or deletions)。分析原理是,以一組" 正常 "細胞作為參考(通常以免疫細胞作為 reference),分析腫瘤基因組上各個位置的基因表達量強度變化。通過熱圖的形式展示每條染色體上的基因相對表達量,相對于正常細胞,腫瘤基因組總會過表達或者低表達。相關文章 2014 年就發表在了 Science 上 [1],之后算法不斷優化,分析結果也多次刊登在 CNS 文章上,所以該算法的可靠性還是有頂刊背書的,下圖是官方給到的分析流程。
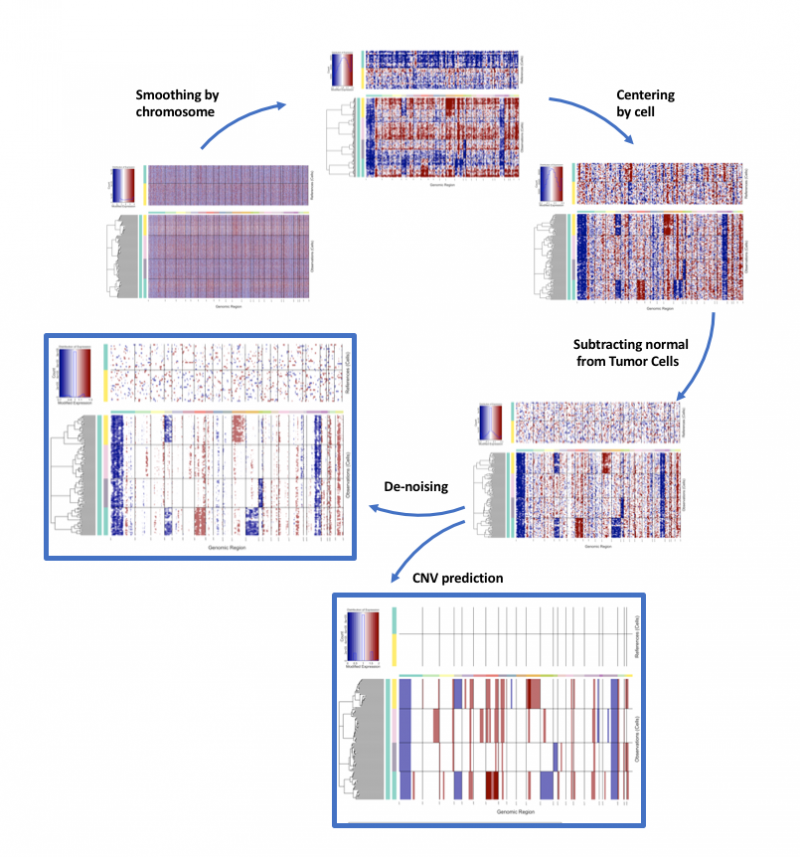
InferCNV procedure
2020 年 7 月 20 日,中山大學腫瘤防治中心、深圳華大生命科學研究院、中山大學生命科學學院與中國科學院深圳先進技術研究院等單位合作在 Cell Research(IF=20.5)雜志上在線發表的鼻咽癌(NPC)研究新成果中 [2],使用 infer CNV 分析對細胞進行核型,并與 WES 數據一致。分別對正常樣本和 15 個腫瘤樣本中的上皮細胞的 CNV 圖譜進行了分層聚類,腫瘤樣本中整個染色體缺失或擴增的細胞被鑒定為惡性細胞,11 個腫瘤樣本中鑒定出 7581 個惡性細胞,并進行后續分析。

文章中 inferCNV 結果
inferCNV 結果里面的 CNV 熱圖,雖然說可以肉眼簡單看看不同的細胞亞群是否具有大面積的 CNV 事件來判定它是否是惡性細胞,但是這樣的判定具有很大程度的主觀性,所以理論上需要有更好的方法,也就是計算具體每個細胞的 CNV score。

scCancer 原理同樣是分析每個細胞基因表達值跟參考 ref(數據庫自帶的 reference)相比的 cnv 信息,并根據整體基因拷貝數變化的情況對細胞進行 cnv 打分(malignScore)。針對癌癥的高度異質性和復雜微環境整合了一組適用于腫瘤數據的計算和分析方法,包括腫瘤微環境分析、惡性細胞評估、細胞周期評估、干細胞特征評估、基因集信息得分評估、表達程序識別、細胞間相互作用分析。此外,這種方法也適用于多樣本整合分析,可以有效消除批次效應提高數據分析結果可靠性。
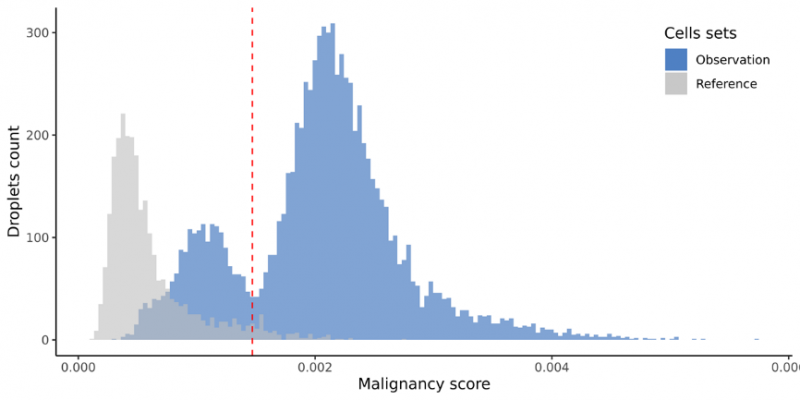
根據 malignScore 峰圖分布計算閾值,超過閾值認為是惡性細胞
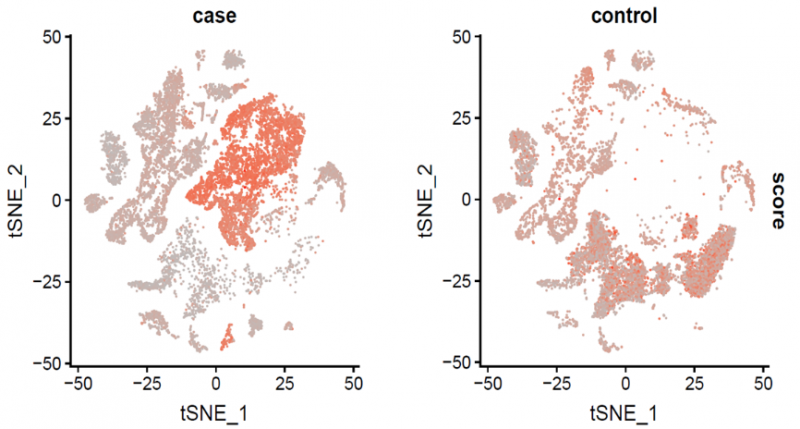

malignScore 分布及分類展示
還有一些研究者使用指定基因的表達量來判斷細胞的良惡性 [3],具體采用哪種分析方法還要根據樣本及數據的具體情況來定,當然也可以多做方法結合。
參考文獻
[1]Patel Anoop P,Tirosh Itay,Trombetta John J et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma.[J] .Science, 2014, 344: 1396-401.
[2] Chen Yu-Pei,Yin Jian-Hua,Li Wen-Fei et al. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma.[J] .Cell Res, 2020, 30: 1024-1042.
[3] Zhang Peng,Yang Mingran,Zhang Yiding et al. Dissecting the Single-Cell Transcriptome Network Underlying Gastric Premalignant Lesions and Early Gastric Cancer.[J] .Cell Rep, 2020, 30: 4317.
更多伯豪生物人工服務:

