隨著測(cè)序技術(shù)的飛速發(fā)展,單細(xì)胞轉(zhuǎn)錄組測(cè)序技術(shù)也已成為實(shí)驗(yàn)室常規(guī)工具之一。然而,研究人員在試圖應(yīng)用單細(xì)胞轉(zhuǎn)錄組技術(shù)的時(shí)候也面臨著令人困惑的選擇,比如說選擇哪種建庫(kù)測(cè)序平臺(tái),使用哪種分析方法以及后續(xù)的生物信息學(xué)分析方法的選擇等等。
此前,來自人類細(xì)胞圖譜聯(lián)盟的研究人員進(jìn)行了一項(xiàng)綜合性多中心研究,通過使用包含人類、小鼠和狗細(xì)胞的參考樣本,比較了 13 種單細(xì)胞轉(zhuǎn)錄組測(cè)序流程的異同。結(jié)果發(fā)現(xiàn)不同流程在量化基因表達(dá)和識(shí)別細(xì)胞類型層面存在著顯著差異。
近日,美國(guó)羅馬琳達(dá)大學(xué)基因組學(xué)中心的研究團(tuán)隊(duì)在 Nature Biotechnology 發(fā)表了題為“A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples”的研究性文章,研究人員設(shè)計(jì)了一項(xiàng)綜合性的多中心研究,用以評(píng)估技術(shù)平臺(tái)、樣品組成和生物信息學(xué)方法(包括預(yù)處理、歸一化和批次效應(yīng)校正)的影響,并在后為科研人員解決科學(xué)問題的技術(shù)平臺(tái)和生物信息方法的結(jié)合提供了實(shí)踐指導(dǎo)。

文章發(fā)表在 Nature Biotechnology
該研究使用了四種測(cè)序平臺(tái):10x Genomics,F(xiàn)luidigm C1, Fluidigm C1 HT 和 Takara Bio ICELL8;測(cè)序工作分別由四個(gè)研究中心完成:Loma Linda University(LLU),the National Cancer Institute(NCI),the US Food and Drug Administration(FDA)和 Takara BioUSA(TBU)。樣本層面,他們使用了有兩個(gè)特征明顯的參考細(xì)胞系:來自同一供者的乳腺癌細(xì)胞系(樣本 A)和“正常”B 淋巴細(xì)胞系(樣本 B)。然后使用 3 '或全長(zhǎng)單細(xì)胞轉(zhuǎn)錄組測(cè)序方法對(duì) 30,693 個(gè)單細(xì)胞進(jìn)行了測(cè)序,共生成了 20 個(gè)數(shù)據(jù)集。
針對(duì)產(chǎn)生的這 20 個(gè)數(shù)據(jù)集,研究人員對(duì)不同的數(shù)據(jù)預(yù)處理方法、數(shù)據(jù)標(biāo)準(zhǔn)化方法、批次效應(yīng)矯正方法等進(jìn)行了評(píng)估。
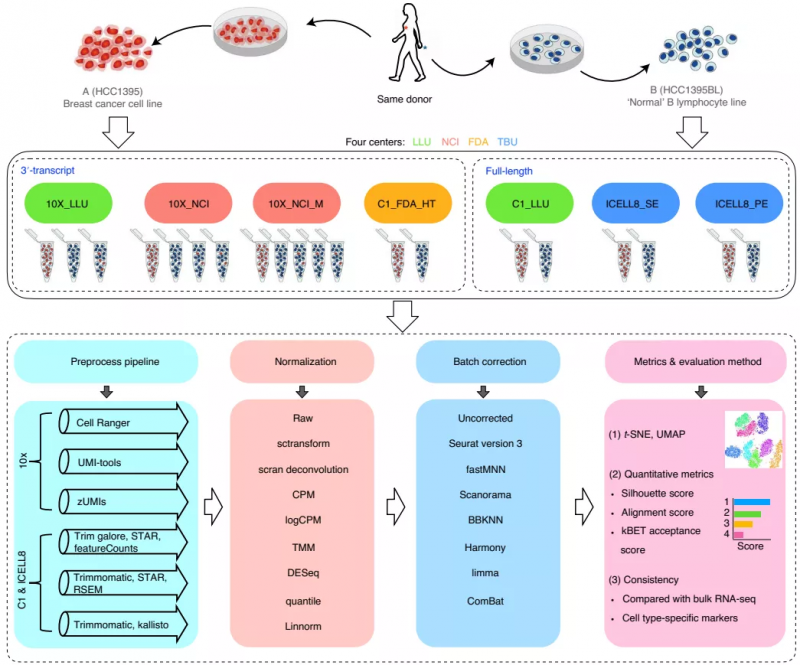
圖 1. 研究總體設(shè)計(jì)示意圖。來源:Nature Biotechnology
測(cè)序深度與檢測(cè)基因數(shù)的關(guān)系
首先,研究人員對(duì)序列深度與檢測(cè)到的基因數(shù)量的關(guān)系進(jìn)行了評(píng)估。正如預(yù)期的那樣,隨著測(cè)序深度的增加,檢測(cè)到的基因數(shù)逐漸升高并趨于穩(wěn)定。另外,對(duì)于癌細(xì)胞(樣本 A)和 B 淋巴細(xì)胞(樣本 B),隨著測(cè)序深度的增加,每個(gè)細(xì)胞檢測(cè)到的基因數(shù)量迅速增加,特別是 Fluidigm C1 平臺(tái)。然而,對(duì)于全長(zhǎng)測(cè)序技術(shù)(C1_LLU 和 ICELL8),在 10 萬(wàn)次讀取后,飽和速率較慢,在相同的測(cè)序深度增加情況下,與基于 3’的測(cè)序技術(shù)相比,額外能夠檢測(cè)到的基因較少。
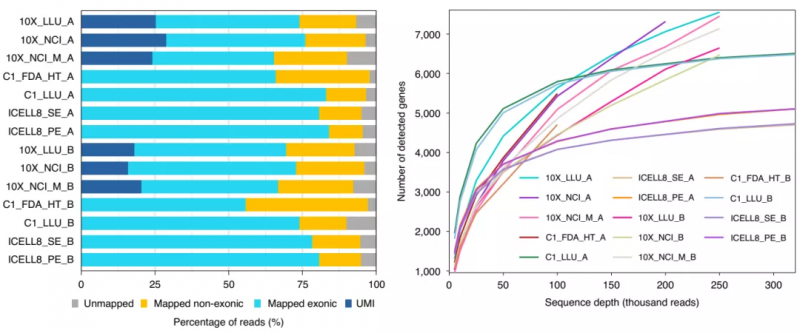
圖 2. 不同測(cè)序平臺(tái)檢測(cè)的基因數(shù)及與測(cè)序深度的關(guān)系。來源:Nature Biotechnology
數(shù)據(jù)預(yù)處理方法的比較
對(duì)基于 UMI(Unique Molecular Identifier)的單細(xì)胞轉(zhuǎn)錄組測(cè)序數(shù)據(jù),他們比較了三種預(yù)處理方法:Cell Ranger 3.1(10x Genomics)、UMI-tools 和 zUMIs。結(jié)果顯示,三種方法在識(shí)別細(xì)胞數(shù)量和每個(gè)細(xì)胞檢測(cè)到的基因數(shù)量層面都存在差異。不過,Cell Ranger V3 是靈敏的細(xì)胞條形碼識(shí)別方法,UMI-tools 和 zUMIs 可以過濾大多數(shù)低基因或轉(zhuǎn)錄表達(dá)的細(xì)胞,但每個(gè)細(xì)胞內(nèi)可檢測(cè)到更多的基因。
對(duì)非基于 UMI 的單細(xì)胞轉(zhuǎn)錄組測(cè)序數(shù)據(jù),他們比較了另外三種預(yù)處理方法:featureCounts、kallisto 和 RSEM。這些數(shù)據(jù)預(yù)處理流程包括去除低質(zhì)量測(cè)序數(shù)據(jù)、基因組比對(duì)和基因計(jì)數(shù)。結(jié)果表明,三個(gè)不同的預(yù)處理方法檢測(cè)到的基因數(shù)量的差異比較大。kallisto 在全長(zhǎng)轉(zhuǎn)錄組測(cè)序數(shù)據(jù)中發(fā)現(xiàn)了每個(gè)細(xì)胞中更多的基因。此外,基于 Fluidigm C1 HT 3’測(cè)序方法產(chǎn)生的數(shù)據(jù)中,kallisto 方法檢測(cè)到的每個(gè)細(xì)胞的基因數(shù)與其它兩個(gè)管道生成的基因序列有顯著差異。
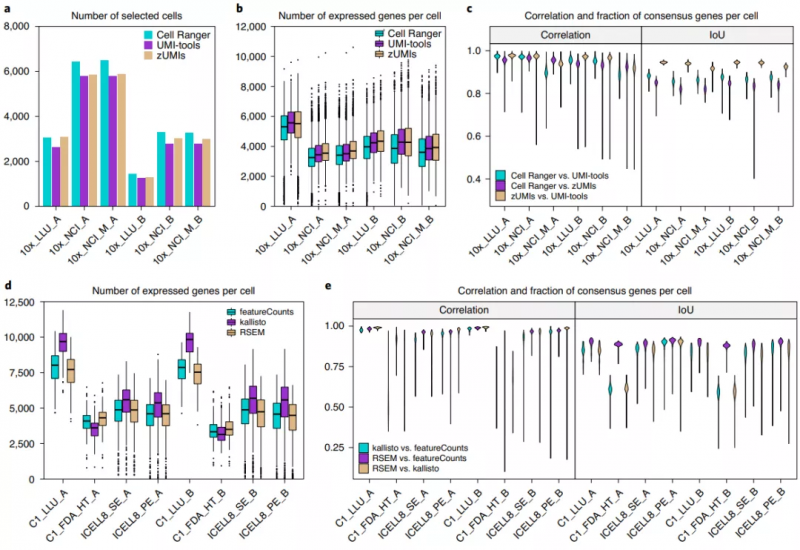
圖 3. 數(shù)據(jù)預(yù)處理方式對(duì)檢測(cè)到的基因數(shù)量的影響。來源:Nature Biotechnology
不同批次矯正算法的比較
如上所述,數(shù)據(jù)集之間的差異可能來自技術(shù)層面或生物因素,針對(duì)這些技術(shù)層面帶來的差異,在進(jìn)行數(shù)據(jù)分析時(shí)是需要矯正的,否則將會(huì)影響結(jié)論。研究者對(duì)七種校正批次效應(yīng)的算法進(jìn)行基準(zhǔn)測(cè)試:Seurat version 3、fastMNN、mutual nearest neighbors(MNN)、Scanorama、BBKNN、Harmony、limma 和 ComBat。
他們通過四種不同的樣本組合評(píng)估這些算法的性能,組合 1 包含所有單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)集,包括混合和純合數(shù)據(jù)集;組合 2 只包含了乳腺癌細(xì)胞系數(shù)據(jù);組合 3 分別對(duì) B 細(xì)胞系來源數(shù)據(jù)進(jìn)行評(píng)估;組合 4 中,數(shù)據(jù)由將 5% 或 10% 的乳腺癌細(xì)胞(樣本 A)加入到 B 淋巴細(xì)胞(樣本 B)中,用 10x Genomics 平臺(tái)橫跨兩個(gè)中心測(cè)序得到。
結(jié)果顯示,在去除批次效應(yīng)和從 B 淋巴細(xì)胞中分離乳腺癌細(xì)胞方面,BBKNN、fastMNN 和 Harmony 是有效的;Seurat V3 是將不同批次的相似細(xì)胞聚集在一起的方法之一,特別是對(duì)乳腺癌細(xì)胞,但也會(huì)存在過度校正的現(xiàn)象,比如將兩種高度不同的細(xì)胞類型融合在一起。另外,當(dāng)只分析來自 10x 平臺(tái)的數(shù)據(jù)時(shí),Scanorama 既能清晰地分離不同的細(xì)胞,又能很好地將相似的細(xì)胞組合在一起。
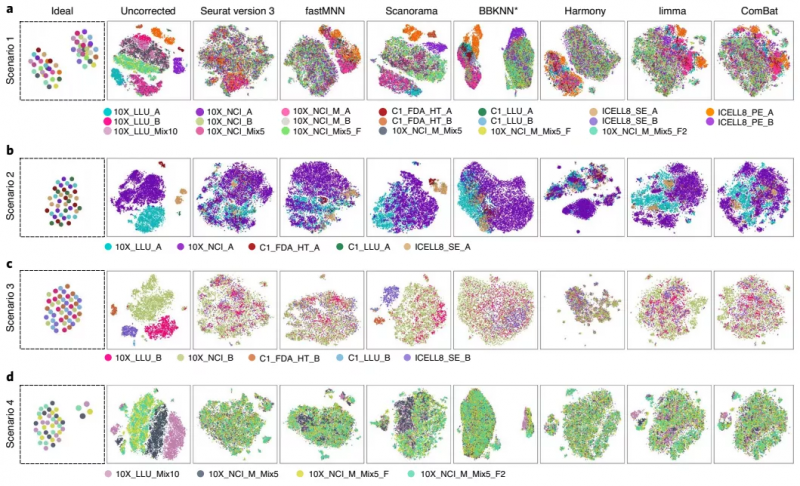
圖 4. 比較分析不同工具的批次矯正效果。來源:Nature Biotechnology
綜合上述的分析結(jié)果,研究人員對(duì)這些預(yù)處理方法和算法進(jìn)行了綜合排序,如圖 5 所示,基于 UMI 的數(shù)據(jù)可以用文中所列的任何方法進(jìn)行預(yù)處理,而 kallisto 則更適用于全長(zhǎng)轉(zhuǎn)錄組測(cè)序數(shù)據(jù)的預(yù)處理。
在跨中心數(shù)據(jù)集,特別是當(dāng)數(shù)據(jù)集中存在大量不相似細(xì)胞時(shí),BBKNN 表現(xiàn)先進(jìn),而 limma 和 ComBat 在兩種類型的細(xì)胞的跨平臺(tái)、跨中心分離中表現(xiàn)差。Seurat V3、fastMNN 和 Harmony 都能很好地混合來自不同平臺(tái)和位點(diǎn)的生物相同或相似樣本的單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)。
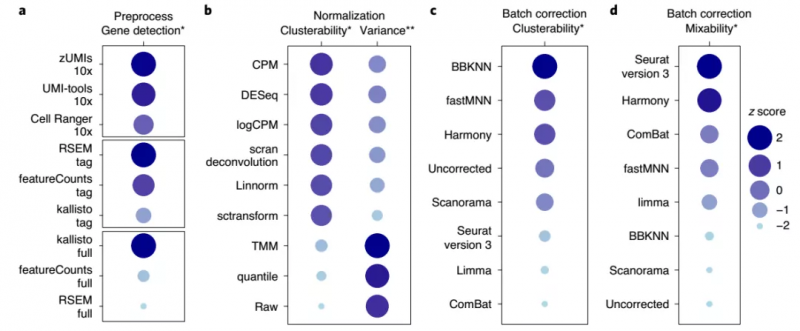
圖 5. 生物信息學(xué)指標(biāo)的性能排名。來源:Nature Biotechnology
綜上所述,該研究比較分析了 6 種單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)預(yù)處理流程、8 種歸一化方法和 7 種批次校正算法,結(jié)果表明,單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)之間的確存在批次效應(yīng),不過,跨中心和不同平臺(tái)的數(shù)據(jù)差異可以通過適當(dāng)?shù)挠?jì)算方法進(jìn)行糾正。同時(shí),該研究也強(qiáng)調(diào)了選擇適合的測(cè)序技術(shù)平臺(tái)和分析數(shù)據(jù)算法的重要性。如下圖所示,他們也根據(jù)本研究結(jié)果為科研人員選擇適合解決科學(xué)問題的技術(shù)平臺(tái)和生物信息方法的結(jié)合提供了實(shí)踐指導(dǎo)。
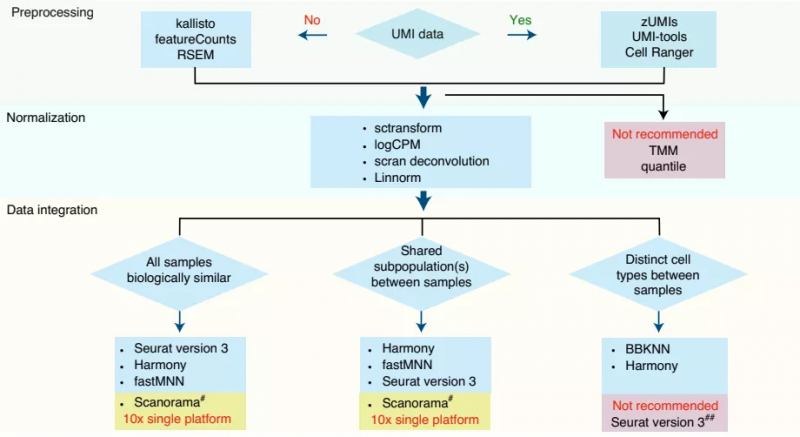
圖 6. 好的分析推薦方案。來源:Nature Biotechnology
參考文獻(xiàn):
1.Chen, W., Zhao, Y., Chen, X. et al. A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples. Nat Biotechnol (2020).
2.Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch efects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
3.Butler, A., Hofman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across diferent conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
轉(zhuǎn)載:測(cè)序中國(guó)(侵刪)

更多伯豪生物人工服務(wù):

